Peptide Synthesis
This article is providing non-experts with strategic considerations and general information about principles of peptide chemistry. The article is not encyclopedic but rather devoted to the history and bases of peptide synthesis.
Brief History of Peptide Synthesis
A «peptide synthesis» includes a large range of techniques and procedures that enable the preparation of materials ranging from small peptides to large proteins. The pioneering work of Bruce Merrifield, which introduced solid phase peptide synthesis (SPPS), dramatically changed the strategy of peptide synthesis and simplified the tedious and demanding steps of purification associated with solution phase synthesis. Moreover, Merrifield's SPPS allows development of automation and the extensive range of robotic instrumentation. After defining a synthesis strategy and programming the amino acid sequence, machines can automatically perform all synthesis steps to prepare multiple peptide samples. SPPS has now become the method of choice to produce peptides, although solution phase synthesis can still be useful for large-scale production of given peptide.
Peptide chemistry is a sub-discipline of chemistry, developed with considerable delay. Its growth is not comparable to the rapid blossoming of aromatic compounds' chemistry which started with the isolation of benzene from coal gas by Faraday in 1825 and the proposition of its structure by A.Kekule in 1865. Those discoveries built a basis of dyes and medicines manufacture and later followed by petro-chemistry production. The prehistory of peptide chemistry lies hidden in early studies of proteins and in physiological chemistry, e.g. finding a relationship between nutrients and the blood compositions.
The first peptide synthesis, as well as a term «peptide», was reported by Hermann Emil Fischer and Ernest Fourneau in 1901. Fischer and E. Fourneau published preparation of first dipeptide, glycylglycine by partial HCl hydrolysis of glycine diketopiperazine.
At German Scientists and Physicians Society, in Karlsbad in 1902, Emil Fischer summarized the isolation of amino acids and peptides from protein hydrolyzates and discussed the coupling of amino acids in proteins. In his own words (translated from German): «the idea that acidamide - like groups play the principal role comes readily to mind, as Hofmeister also assumed in his general lecture this morning». Fischer had begun experiments to link amino acids to each other by methods of organic chemistry as early as 1900. The two lectures mark the emergence of the so called Fischer-Hofmeister theory of protein structure. Emil Fischer, who in a paper with E. Foumeau had just described the first prototype, glycyl-glycine, proposed in his lecture also the designations «peptide, dipeptide, tripeptide etc.» and later «polypeptides».
Emil Fischer and his co-workers synthesized about 100 peptides within the first decade of the 20th century using the methods developed in his laboratory, peptides containing from 2 to 18 amino acid residues.
Synthetic work with peptides was resumed by Bergmann along the lines of the Fischer school in a systematic way with Joseph S. Fruton, who joined Bergmann in 1934, soon after his arrival at the Rockefeller Institute. Fruton utilized the potential of the new carbobenzoxy method to synthesize inaccessible peptides for testing as substrates for enzymatic hydrolysis. He and Bergmann published a syntheses of simple substrates which were cleaved by papain, chymotrypsin, trypsin, and pepsin at certain peptide bonds.
The history of peptide chemistry stretches over a century, from compounds as simple as diglycine to enzymes with more than a hundred amino acids. Due to the individual character of each amino acid and combination of many amino acids forming large molecules, peptides show an extremely great variety of specific functions. They can act as chemical messengers, hormone, intra- or inter-cellular mediators, highly specific stimulators and inhibitors and as biologically active peptides in the brain and nervous systems. Many antibiotic compounds from bacteria, molds and amphibian skin are peptic in nature and so are compounds toxic not only for pathogens but also for cells of higher organisms. Peptides actively involved in reactions of the immune system are attracting increasing attention from biomedical researchers. Peptide science is still in full development.
Protective Groups for Peptide Synthesis
Because of amino acid is an acid with a basic group at one end and an acid group at the other, polymerization of amino acids is common in reactions where each amino acid is not protected. In order to prevent this polymerization, protective groups are used.
Currently, two protecting groups are commonly used in solid-phase peptide synthesis – Fmoc (9-fluorenylmethyl carbamate) and t-Boc (Di-tert-butyl dicarbonate).
Fmoc Protecting Group
The use of Fmoc chemistry for protection of the alpha amino group has become the preferred method for most contemporary solid and solution phase peptide synthetic processes. Fmoc has also been shown to be more reliable and produce higher quality peptides than Boc chemistry.
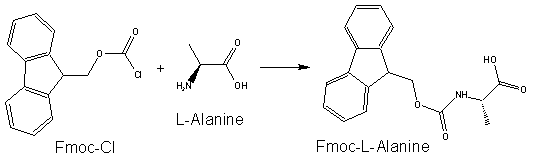
The advantage of Fmoc is that it is cleaved under very mild basic conditions (e.g. piperidine), but stable under acidic conditions. After base treatment, the nascent peptide is typically washed and then a mixture including an activated amino acid and coupling co-reagents is placed in contact with the nascent peptide to couple the next amino acid. After coupling, non-coupled reagents can be washed away and then the protecting group on the N-terminus of the nascent peptide can be removed, allowing additional amino acids or peptide material to be added to the nascent peptide in a similar fashion.
Boc Protecting Group
Before the Fmoc group became popular, the t-Boc group was commonly used for protecting the terminal amine of the peptide, requiring the use of more acid stable groups for side chain protection in orthogonal strategies. Boc groups can be added to amino acids with Di-tert-butyl dicarbonate (Boc anhydride) and a suitable base.
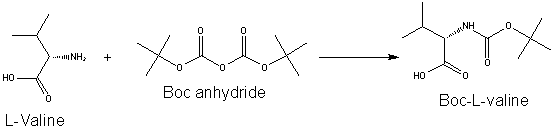
The t-Boc protecting group is removed by exposing the Boc-protected residue on the chain to a strong acid. Typical reagents of choice for deprotection in existing methods are trifluoroacetic acid (TFA) in dichloromethane, hydrochloric acid or methanesulfonic acid in dioxane. The acid used to remove the Boc protecting group is typically neutralized with a tertiary amine such as N-methylmorpholine, N-diisopropylethylamine (DIEA) or triethylamine (TEA).
Basic principles and reagents in peptide synthesis
A peptide bond formation requires an activation of amino acid carboxyl group,
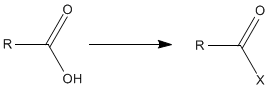
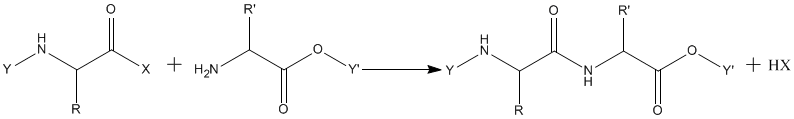
The protection groups (Y) were not meant to be part of amides and remained problematic for a long time. The first peptide derivatives secured by synthesis, benzoylglycyl-glycine and ethoxycarbonylglycyl-glycine ethyl ester carried blocking groups which could not be removed without the destruction of a newly formed peptide bond. It became obvious that for peptide synthesis easily removable protecting groups were needed. A breakthrough toward the solution of this problem was discovery of benzyloxycarbonyl (carbobenzoxy) group (Z or Cbz) by remarkably lasting contribution of Bergmann and Zervas in 1932.
The benzyloxycarbonyl group can be removed by catalytic hydrogenation, at room temperature and ordinary pressure, a process that leaves the peptide bond and the various side chain functions unaffected and generates harmless byproducts, toluene and carbon dioxide.
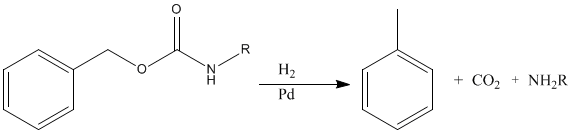
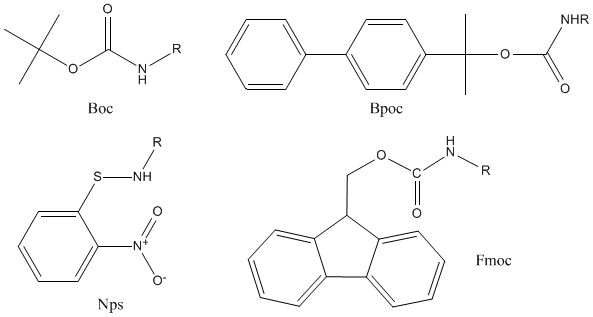
A removal of Z group stimulated further research toward acid-sensitive protecting groups. This led to development of series of blocking groups cleavable under mild conditions.
The most important were for instance, tert-butyloxycarbonyl (Boc), O-nitrophenylsulfenyl (Nps) and biphenylylisopropyloxycarbonyl (Bpoc) groups. De-protection by nucleophiles appears to be an attractive alternative to acidolysis in case of Nps group. Cleavage of 9-fluorenylmethyloxycarbonyl (Fmoc) group by secondary amines gained considerable significance in the synthesis of complex peptides.
Coupling through anhydrides, however, became popular among peptide chemists. Due to variability of the components used for activation of protected amino acids, mixed or unsymmetrical anhydrides, were proposed by many investigators as different versions of general approach to peptide bond formation.
The yields and purity of peptides may have been reached by using isopropyl, isobutyl or sec-butyl carbonic anhydrides.
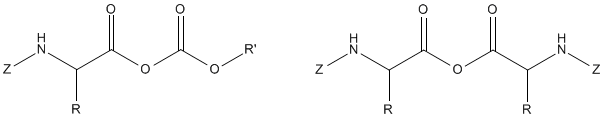
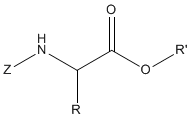
Some esters became very popular, e.g. nitrophenyl ester, 2,4,5-trichlorophenyl ester, N-hydroxysuccinimide ester and pentaftuorophenyl ester.
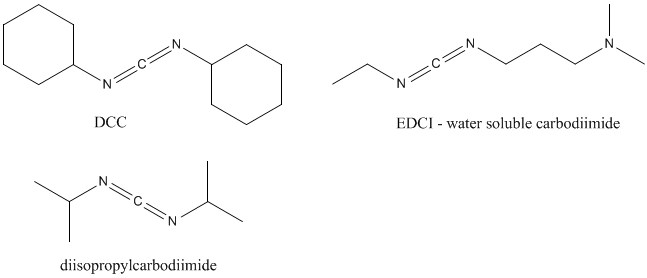
The «coupling reagents» are used in peptide synthesis as attractive peptide bond formation method. These are compounds which can be added to reaction mixture of both a partially protected amino acid or a peptide with a free carboxyl group and the second protected amino acid or peptide with a free amino group. The most successful coupling reagent, dicyclohexylcarbodiimide (DCC or DCCI), was introduced by Sheehan and Hess in 1955. DCC, its water soluble derivatives (EDCI; 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide) and diisopropylcarbodiimide are lead coupling reagents till now.
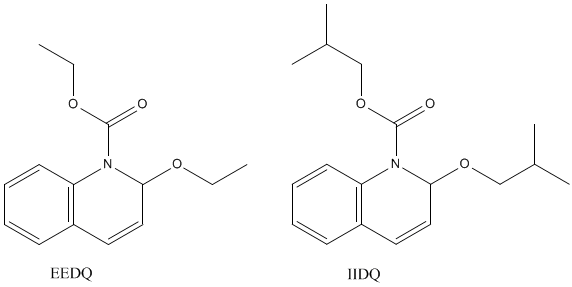
The mixed-anhydride-producing compound 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) and an analog - 1-isobutoxycarbonyl-2-isobutoxy-1,2-dihydroquinoline (IIDQ), are useful coupling reagents. They are readily prepared, easily stored and cause little racemization or other side reactions.
Most of the peptide synthesis are accompanied by minor, undesired side reactions, and the peptides can be contaminated by by-products. There's a solution that avoids by-products formation.
Solid-Phase Peptide Synthesis
Peptide synthesis is much more complicated than simply forming amide bonds by mixing the desired amino acids in a reaction vessel. Twenty natural and endless number of unnatural amino acids give numerous possible combinations formed with this technique.
In a solution of two amino acids mixed together, four different dipeptides as well as other longer peptides will be formed, e.g. in a mixture of glycine and alanine four dipeptides would be Gly-Gly, Gly-Ala, Ala-Gly and Ala-Ala.
The solid-phase peptide synthesis was developed for the purpose of providing rapid, simplified and effective way of peptides and small proteins preparation (Merrifield, 1963).
To ensure the desired peptide is formed, the basic group of amino acid and the acidic group of another must both be made unable to react. Such «deactivation» is known as the protection of reactive groups. A group that is unable to react, is called a protecting group.
In classical organic synthesis the acids are protected, allowed to react and de-protected. Afterward one end of peptide is protected and allowed to undergo a reaction with a new protected acid and so on. In solid-phase peptide synthesis (SPPS) one peptide end is attached to water-insoluble polymer and remains protected throughout the entire peptide formation, meaning both fewer steps and simplified purification, the reagents can be rinsed away without losing the peptide.
Solid-phase peptide synthesis involves the following steps: attaching amino acid to the polymer, protection, coupling, de-protection and polymer removal.
Attaching of amino acid to polymer
The attachment is done by reaction of amino acid with a linkage agent and following reaction of linkage another end with a polymer. A peptide-polyamide link is formed and will not be hydrolyzed during the subsequent peptide-forming reactions. Common linkage agents are di- and tri-substituted benzenes as shown below.
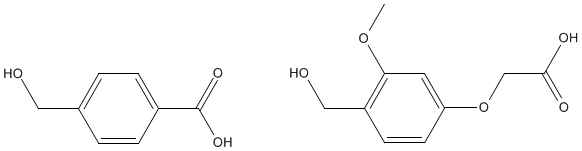
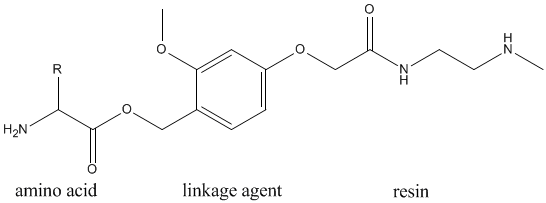
These together with the C-terminus amino acid and resin form the following polymer structure.
Protection
The next amino acid needs to have its amino group protected to prevent the acids reacting with each other. FMOC (9-fluorenylmethoxycarbonyl) is used as an excellent protecting group. Moreover, any amino acid side chains containing aromatic, acid, basic and/or polar groups are likely to be reactive. They must be protected to prevent undesired branched chains from forming. There are four main groups used on this way, tertiary butyl group (tBu), triphenylmethyl group (Trt), tertiary butyloxycarbonyl group (tBOC) and 2,2,5,7,8-pentamethylchroman-6-sufonyl group (PMC).
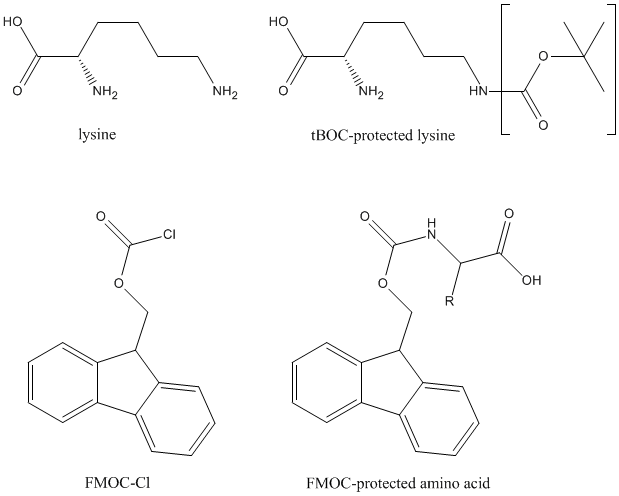
Coupling
The FMOC protected amino acid reacts with the last amino acid attached to the polyamide. The reaction is catalyzed by DCC (N,N'-Dicyclohexylcarbodiimide).
De-protection
DCC excess is washed off by water. FMOC group is removed by piperidine (secondary cyclic amine). The acid protection group is removed from the chain and is able to add another acid molecule. On the step two a new amino acid is protected. The cyclic process continues until a required chain length has been accomplished.
Polymer removal
Once the desired peptide is completed it must be removed from polyamide. It can be reached by using 95% solution of trifluoroacetic acid (TFA). The side-chain protecting groups are removed at this stage as well.
Advantages and disadvantages of SPPS
The main advantage of SPPS is a peptide high yield. As peptide consists of many amino acids and each acid yield is less than 100%, overall peptide yield is negligible. For example, if each amino acid gives 90% an overall yield of peptide containing 100 amino acid is 0,003% only.
Solid-phase peptide synthesis is much faster than classical solution process. SPPS allows a formation of 20 amino acids peptide in 24 hours and longer ones in less than a week.
As modern automated synthesizers and sophisticated analytical and purification equipment are improved, a peptide chemist can make peptides in the range of 20-50 amino acids in length and in amounts from 20-100 milligrams.
The essential advantage of SPPS is efficiency of separation of the intermediates from starting materials, reagents and most of the by-products. The peptide, attached to the insoluble polymer, remains undissolved during solvents' treatment.
Another plus of SPPS is absence of time consuming isolation of intermediates through extraction or crystallization. A problem to find solvents for large molecular weight intermediates no longer exists because it's sufficient using solvents in which polymer just swells.
It becomes rather simple to convert amino salt to free amine, by treating peptidyl resin with tertiary base and following removal of alkylammonium salt formed, by washing.
The SPPS biggest weakness: the intermediates cannot be purified and the possibilities for their characterization are rather limited.
However, numerous instruments for the automatic execution of SPPS have been described and several methods were developed for the continuous analytical control of peptide building.
References
1. M. Bodanszky. Principles of Peptide Synthesis, 2nd ed., Springer, Heidelberg, 1993. [Book].
2. Bernd Gutte. Peptides: Synthesis, Structures, and Applications. Academic Press; 1 edition (November 2, 1995). [Book].
3. Gregg B. Fields. Introduction to Peptide Synthesis. Current Protocols in Protein Science. [Article].
4. T. Wieland, M. Bodanszky. The World of Peptides. A Brief History of Peptide Chemistry. [Book].
5. Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int J Pept Protein Res. 1990; 35:161–214. [Article].
6. Merrifield B. Solid phase synthesis. Science. 1986; 232:341–347. [Article].